Hepatic encephalopathy
Hepatic encephalopathy or portosystemic encephalopathy is a syndrome of neuropsychiatric dysfunction caused by portosystemic venous shunting, with or without liver disease, but primarly occurring in patients with advanced liver failure.
Pathogenesis:
Ammonia is the primary neurotoxin that precipitates hepatic encephalopathy. The increase in blood ammonia in advanced liver disease is a consequence of impaired liver function with shunting of blood around the liver. Muscle wasting, a common occurrence in these patients, also may contribute since muscle is an important site for extrahepatic ammonia removal.
Ammonia Metabolism:
Ammonia metabolism involves primarily five organs—the gut, kidney, muscle, liver, and brain. Ammonia is produced mostly in the gut, but also in the kidney and muscle.
The gastrointestinal tract is the primary source of ammonia, which enters the circulation via the portal vein. Ammonia is produced by enterocytes from glutamine and by colonic bacterial catabolism of nitrogenous sources, such as ingested protein and secreted urea. Within the kidney, ammonia is produced and is essential for the renal handling of acid. Renal ammonia production is dynamic and increases with alterations in renal acid-base status changes and with GI bleeding. Skeletal muscle can also produce ammonia, usually during seizures or with intense exercise. The intact liver clears almost all of the portal vein ammonia, converting it into urea or glutamine and preventing entry into the systemic circulation.
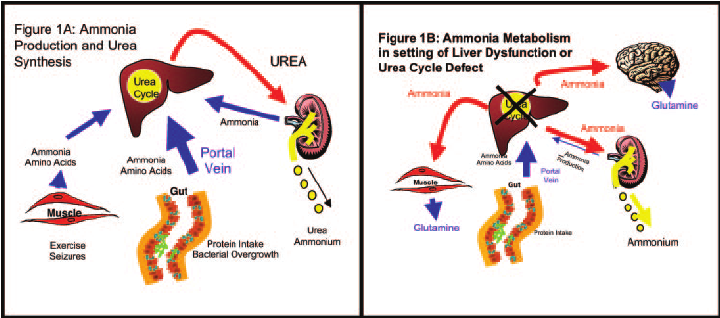
The liver is primarily responsible for ammonia degradation. Ammonia in the venous system (which is produced by the digestion of protein in the splanchnic circulation and by muscle peripherally) is metabolized to urea through the urea cycle. When the capacity of the liver to metabolize ammonia is overcome, either because ammonia production exceeds the metabolic capacity of the liver or because the liver is unable to metabolize ammonia, elimination is dependent on the kidney, muscle, and brain.
In the setting of hyperammonemia, the kidney decreases ammonia production and increases urinary excretion of ammonia. The muscle and brain metabolize excess ammonia to glutamine. The process of metabolizing ammonia to glutamine is physiologically costly, particularly in the brain where the symbiotic relationship between neurons and astrocytes is disrupted by excess glutamine production. Astrocytes rapidly metabolize ammonia to glutamine, but the subsequent rise in intracellular osmolarity causes astrocyte swelling and cerebral edema. Cerebral edema and herniation (as well as seizures) are unique to acute hyperammonemia and usually occur when arterial ammonia levels are >200.
Clinical features:
Patients with grade I encephalopathy may have mild asterixis, whereas pronounced asterixis is seen in patients with grade II or III encephalopathy. Asterixis is typically absent in patients with grade IV encephalopathy, who instead may demonstrate decorticate or decerebrate posturing.
Cognitive findings in patients with hepatic encephalopathy vary from impairments in attention, reaction time, and working memory to hepatic coma. Neuromuscular impairments include bradykinesia, hyperreflexia, rigidity, myoclonus, and asterixis.
Diagnosis
-
Increased ammonia levels. While ammonia levels correlate with the severity of hepatic encephalopathy, levels are inconsistently elevated. While arterial and venous ammonia concentrations are often elevated in patients with hepatic encephalopathy, an elevated ammonia level is not required to make the diagnosis. In addition, elevated ammonia levels may be seen in patients who do not have hepatic encephalopathy.
-
A single normal ammonia level does not rule out hepatic encephalopathy in a patient with chronic liver disease.
-
Venous ammonia concentration is not useful for screening for hepatic encephalopathy since levels may vary.
-
Serum ammonia levels should not be used to screen for hepatic encephalopathy in patients who are asymptomatic or who have mental status changes in the absence of liver disease or a portal-systemic shunt.
-
CT scan of the brain to r/o subdural hematoma; a CT scan may also demonstrate cerebral edema.
-
EEG should be considered in the presence of focal neurologic signs or seizure activity, or in a comatose patient.
-
Routine tests like CBC,CMP, Blood cultures,USG of liver to look for precipitating causes
-
Monitor hemodynamics and fluid status to look for precipitating factors
-
Possible precipitating causes of the hepatic encephalopathy are:
-
GI bleeding
-
Infections
-
Hypovolemia
-
Hypoxia
-
Sedatives or tranquilizers
-
Hypokalemia and/or metabolic alkalosis potently stimulates renal ammoniagenesis
-
Renal failure including the hepatorenal syndrome may decrease renal ammonia excretion.
-
Hypoglycemia
-
Constipation
-
Vascular occlusion (hepatic vein or portal vein thrombosis) causes increased portosystemic venous blood shunting
-
TIPS placement
-
Excessive dietary protein
-
Hepatocellular carcinoma
-
Acute deterioration of liver function in cirrhosis
-
Acute liver failure of any cause
-
Treatment:
Treatment includes determining the appropriate setting for care, correcting any predisposing conditions, and lowering blood ammonia levels with medications such as lactulose, lactitol, or rifaximin. Restricting dietary protein is not recommended for the majority of patients.
Agitation: Agitation often resolves with treatment of the hepatic encephalopathy; however, patients may represent a hazard to themselves and their caregivers until treatment takes effect. Management may include judicious use of restraints, which may be a safer option than pharmacologic treatment, since patients with advanced liver disease and hepatic encephalopathy may be particularly vulnerable to oversedation with medications. Should medications be required, haloperidol is a safer option than benzodiazepines. Patients with advanced cirrhosis may be particularly sensitive to benzodiazepines.
Protein restriction and nutritional support — Nutritional support should include maintaining an energy intake of 35 to 40 kcal/kg/day, with a protein intake of 1.2 to 1.5 g/kg/day. Patients with chronic HE occasionally do not tolerate normal amounts of dietary protein. A switch to vegetable protein or supplementation with branched-chain amino acids may be considered in this situation. Branched-chain amino acid preparations are relatively expensive.
Lower blood ammonia — The most important step in the treatment of hepatic encephalopathy is initiation of measures to lower blood ammonia concentrations (whether or not the values are frankly elevated) with medications such as lactulose, lactitol, or rifaximin. Correction of hypokalemia is also an essential component of therapy since hypokalemia increases renal ammonia production.
Lactulose: The dose of lactulose (30 to 45 mL given two to four times per day) should be titrated to achieve 3-4 soft stools per day. In the colon, lactulose and lactitol are catabolized by the bacterial flora to short chain fatty acids (eg, lactic acid and acetic acid), which lower the colonic pH to about 5.0. The reduction in pH favors the formation of the nonabsorbable NH4+ from NH3, trapping NH4+ in the colon and thus reducing plasma ammonia concentrations.
Lactulose cannot be given orally in patients with suspected ileus. The amount of lactulose administered by enemas is much greater than that administered orally or by nasogastric tube.
Rifaximin: For patients who have not improved within 48 hours or who cannot take lactulose or lactitol, rifaximin should be added. The dose of rifaximin is 400 mg orally three times daily or 550 mg orally two times daily. As a general rule, antibiotics are added to, rather than substituted for, lactulose.
Golytely: In a recent study, golytely led to more rapid resolution of hepatic encephalopathy than lactulose therapy. HELP Trial
Without identifiable precipitating factors, persistent hepatic encephalopathy related to deteriorating liver function or after TIPS may require chronic therapy with lactulose or the nonabsorbable antibiotic rifaximin to suppress hepatic encephalopathy. Chronic antibiotic administration can lead to bacterial resistance, bacterial overgrowth, and fungal colonization.
The presence of large spontaneous portal-systemic shunts should be sought in selected patients with recurrent episodes of encephalopathy despite medical therapy, where a precipitating factor is not found. A significant proportion of patients receiving TIPS develop hepatic encephalopathy, with the rate of hepatic encephalopathy dependent on the patient’s residual liver function.
Increased ICP:
Hyperammonemia in adults is associated with cerebral edema, decreased cerebral metabolism, and increases in cerebral blood flow. The increased cerebral flow is to divert circulation from portal system to brain to metabolize ammonia. The management of these patients should focus on the reduction of cerebral edema and cerebral blood flow. Management of elevated ICP can be done by:
-
Mannitol : A bolus of mannitol (0.5 to 1.0 g/kg) is typically the first-line therapy for patients with an elevated ICP. Doses can be repeated every 4 hours or infused as a drip to keep serum osmolarity between 300-320.
-
Hypertonic saline ( 2%, 3% and 23%) : the goal is to keep serum sodium between 145-155. 2% saline can be given from peripheral IV and 3% and 23% saline needs a central line for administration. Hypertonic saline boluses can be given as 150ml of 3% saline or 30 ml of 23% saline.
Sodium needed in mmol = (lean body weight in kg × 0.5 for a woman or 0.6 for a man) × (target sodium − current sodium in mmol/L).Divide the result of this (# of mmol) by the concentration of your NaCl solution (in mmol/L) to get a total volume (L) to bolus in measured aliquots -
Hyperventilation: Decreasing the PaCO2 to 25 to 30 mmHg via hyperventilation results in cerebral vasoconstriction with decrease in cerebral blood flow and a reduction in the ICP. It is always a temporising measure and may be helpful when patients are being transported within institution. Also, in a study, hyperventilation didn't completely improve cerebral edema. J Hepatol. 1986;2(1):43-51
-
Barbiturate coma: it decreases the brain activity and hence, the need for cerebral blood flow. Hence, it could decrease ICP if all measures fail.
-
Craniectomy : If all measures fail, this may be a last resort.