PULMONARY HYPERTENSION
Pulmonary hypertension (PH) is defined simply as a mean pulmonary artery pressure of 25 mmHg or greater. The normal mean pulmonary arterial pressure is 14+-3, with upper limit being 20. Normal pulmonary arterial systolic pressure is around 10 and diastolic pressure around 5 mmHg.
Although pulmonary hypertension is primarly due to elevation of pressure in the pulmonary arterial system alone ( pre-capillary PH), it can also happen secondary to elevations of pressure in the pulmonary venous and pulmonary capillary systems (pulmonary venous hypertension; post-capillary PH), as in left sided heart failure.
Right ventricular failure is a common complication of pulmonary hypertension (PH). Clinically, RV failure is characterized by a reduced cardiac output (i.e., cardiac index<2.5 L/min/m2) and an elevation in RV filling pressure (i.e., right atrial pressure > 8 mm Hg). RV failure can also result from other diseases such as myocarditis, cardiomyopathy, or myocardial infarction.
Although any form of PH can result in RV dysfunction, the full picture of RV failure with low cardiac output and elevated RV filling pressures is typically seen in patients with pulmonary arterial hypertension (PAH) or chronic thromboembolic pulmonary hypertension (CTEPH). On occasion, patients may present with clinical signs of RV failure in a state of markedly elevated rather than reduced cardiac output. This so-called high-output failure is typically seen in patients with large arteriovenous malformations (e.g. hereditary hemorrhagic telangiectasia) , or in patients with chronic hemolytic anemia (e.g., sickle cell disease).
Physiology:
The RV is embryologically, morphologically, and functionally distinct from the left ventricle . After birth it assumes the adult phenotype of a relatively thin-walled, crescent-shaped structure that is adapted to eject into the pulmonary circulation; a circuit characterized by low resistance, high compliance, and low impedance . The RV and LV are interrelated by the shared interventricular septum. The relatedness is also conferred by the surrounding pericardium, which ensures a consistent beat-to-beat intracardiac volume. RV–LV interaction, under normal conditions, allows the ejection of the RV to be augmented by left ventricular ejection . Although highly efficient, the naive RV poorly adapts to sudden increases in afterload . An increase in RV end-diastolic volumes likely initially improves cardiac output by the Frank-Starling mechanism; however, a severe and sudden increase in right ventricular afterload may overwhelm the contractile capability of the RV and lead to hemodynamic collapse.
When RV afterload increases more gradually, RV adaptation occurs. However, in the face of a progressive or sudden worsening in RV afterload these compensatory mechanisms are overwhelmed.
A reduction in oxygen delivery in PAH may be mediated through two mechanisms. First, it may result from a decrease in LV filling resulting from reduction in pulmonary blood flow. Second, RV enlargement may lead to reduced LV filling because of direct RV impingement on LV filling mediated by septal wall motion displacement by a pressure- and volume-overloaded RV. In this instance an elevation in right atrial pressure (a reasonable surrogate for pericardial pressure) should raise the possibility that changes in pulmonary capillary wedge pressure (PCWP) will be an unreliable estimate of LV preload. As such it becomes important for the clinician to consider transmural pressure (left ventricular end-diastolic pressure – pericardial pressure) as a more accurate reflection of LV filling. Practically, transmural pressure may be estimated as PCWP – right atrial (RA) pressure . It is important to recognize that in this adverse situation, volume loading may paradoxically lead to a reduction in left ventricular filling . Conversely, a reduction in right ventricular volume (mediated through diuresis) may improve left ventricular filling and cardiac output through a reduction in pericardial pressure and reduced influence of septal displacement. In this volume overloaded state, both RV filling and LV filling are also highly susceptible to the deleterious effects of tachycardia and tachyarrhythmia, which may further reduce LV filling and stroke volume.
Factors triggering Right Ventricular failure in patients with Pulmonary Hypertension:
Progressive obliteration of the pulmonary vascular bed inevitably results in RV failure once the adaptive mechanisms of the RV are exhausted . Contemporary treatment with endothelin receptor antagonists, phosphodiesterase (PDE)-5 inhibitors, and prostacyclin derivates can sometimes slow disease progression in patients with PAH; however, mortality remains high in this patient population. Hence, RV failure is frequently encountered as a manifestation of disease progression despite targeted therapy. In many instances, however, triggering factors causing or aggravating RV failure can be identified, especially infections, anemia, trauma, surgery, pregnancy, nonadherence with therapy, pulmonary embolism, and arrhythmias. It is likely that the bowel is a major source of bacteremia and endotoxinemia in patients with pulmonary hypertension, as the combination of low cardiac output and elevated venous pressures may result in a loss of the intestinal barrier function.
Arrhythmias are another treatable cause of RV failure in patients with PH. Whereas ventricular arrhythmias, especially ventricular flutter and ventricular fibrillation, have rarely been reported in these patients , atrial tachyarrhythmia (most importantly atrial tachycardia), atrial flutter, and atrial fibrillation are increasingly encountered . As augmented atrial contractility is an important compensatory mechanism in patients with a noncompliant RV, the loss of atrial contractions may have deleterious consequences for RV function. In patients with advanced PAH, new-onset atrial flutter or atrial fibrillation almost invariably results in RV failure. Rate control alone does not appear to be sufficient and restoration of sinus rhythm seems to be critical. Antiarrhythmics or electrical cardioversion may be required when patients are acutely unstable or have a new onset of arrhythmia. In general, b-blocking agents and calcium channel blockers should be avoided as they may further impair RV function. Digitalis glycosides are of limited value but may be used for rate control.
Monitoring in ICU:
Evaluation of cardiac function as well as end-organ function is critical in managing patients with RV failure . Measurements of renal, liver, and neurological function will provide some information about the adequacy of cardiac function and tissue perfusion.
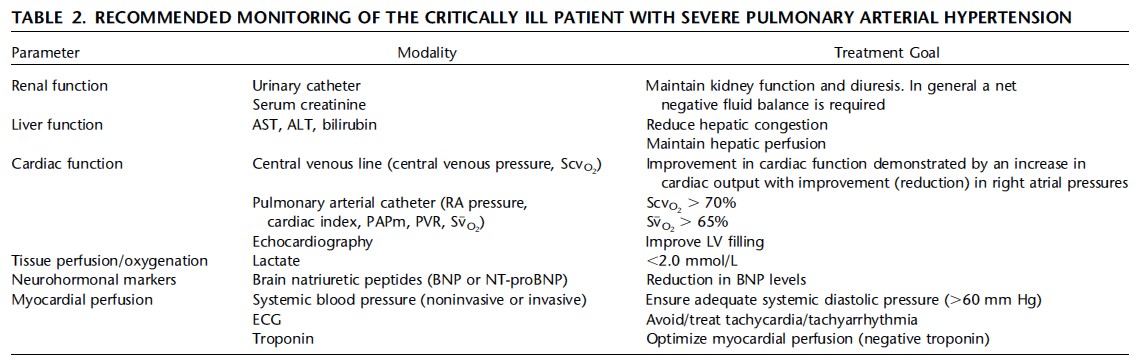
For patients admitted to the ICU with severe PH and RV failure, we advocate for the placement of invasive methods allowing for the measurement of RA pressure, left atrial pressure, cardiac output, and mixed venous oxygen saturation (SvO2). Measurement of pulmonary vascular resistance (PVR) is a composite index of pulmonary pressure and cardiac output. Plasma lactate levels should be monitored closely, as elevated and/or increasing levels may signal progressive RV failure. The use of brain natriuretic peptide measurements to guide care may be of value to document trends in the adequacy of cardiac function over time.
RV failure eventually results in multiorgan dysfunction. Reduced cardiac function can result in deceased bowel perfusion, loss of the intestinal barrier function, and bacterial translocation, a complication that has been implicated as a common cause of death in these patients. Reduced hepatic perfusion can impair liver function or may even result in liver failure. Renal failure is another disastrous complication of RV failure.
Pathogenesis:
PAH is a syndrome resulting from restricted flow through the pulmonary arterial circulation, which leads to pathological increases in PVR and ultimately to right heart failure. The predominant cause of increased PVR is loss of vascular luminal cross section due to vascular remodeling.
The prostanoids, prostacyclin and thromboxane A2, are major arachidonic acid metabolites. Prostacyclin is a potent vasodilator, inhibits platelet activation, and has antiproliferative properties, whereas thromboxane A2 is a potent vasoconstrictor and promotes proliferation platelet activation. In PAH, the balance between these 2 molecules is shifted toward thromboxane A2, favoring thrombosis, proliferation, and vasoconstriction. Additionally, prostacyclin synthase is decreased in the small- and medium-sized pulmonary arteries in PAH.
While RV hypertrophy and dilatation are initiated by increased afterload (i.e., elevated PVR), the adequacy of the RV’s compensatory response (preservation of stroke volume) is quite variable amongst individuals. It remains unclear why some RVs compensate while others decompensate, manifest as thinning and dilatation of the wall, and reduce the RV ejection fraction.
PH in association with left heart failure usually occurs initially as pulmonary venous hypertension. Over a period of time, partly as an adaptive mechanism against pulmonary edema, an increase in pulmonary artery resistance may ensue.
Classification:
-
Pulmonary arterial hypertension (PAH)
-
Idiopathic (IPAH)
-
Familial (FPAH)
-
Congenital heart disease with left to right shunt resulting in increased blood flow
-
Connective tissue disorder like CREST syndromes
-
Portal hypertension causing portopulmonary hypertension
-
Pulmonary veno-occlusive disease (PVOD). Characteristic CT findings are sub pleural thickened septal lines, centrilobular ground-glass opacities (contrasting with a pan lobular distribution found in IPAH), and mediastinal lymphadenopathy. Other findings may include interstitial edema with diffuse central ground-glass opacification and thickening of interlobular septa. Confirmatory test is a lung biopsy.
-
HIV related
-
Drugs and Toxins like cocaine, Amphetamines, SSRI
-
Hemoglobinopathies like sickle cell disease and thalassemia and other hemolytic anemias. (Due to high rate of consumption of NO).
-
Pulmonary hypertension with left heart disease ( Pulmonary Venous Hypertension)
-
Pulmonary hypertension associated with lung diseases and/or hypoxemia
- Chronic obstructive pulmonary disease
- Interstitial lung disease
- Sleep disordered breathing
- Alveolar hypoventilation disorders
- Chronic exposure to high altitude
-
Pulmonary hypertension due to chronic thrombo-embolic disease (CTEPH). Diagnostic test of choice for CTEPH is V/Q scan , as it has higher sensitivity than CT chest. V/Q scan shows multiple segments of perfusion defects.
-
Miscellaneous : Sarcoidosis, histiocytosis X, lymphangiomatosis, ESRD on HD, myeloproliferative diseases, splenectomy, vasculitis, thyroid diseases.
Causes of acute pulmonary hypertension include PE, ARDS, post cardiac surgery, post transplant, COPD exacerbation, pulmonary veno occlusive disease, Acute left sided failure like MR,MS,VSD, LV infarct.
Diagnosis/Screening:
The most appropriate study to obtain in patients suspected of having PH based on history, physical examination, chest x-ray (CXR), electrocardiogram (ECG) and an echocardiogram. Also, BNP and troponins might be elevated and carry poor prognosis.
When PH of any cause is suspected, the best first step is ECHO to estimate the pulmonary artery systolic pressure (PASP) and to evaluate for right ventricular dysfunction, left heart disease, and intracardiac shunt with agitated saline.
-
If the PASP is <35-40 AND there is no evidence of RV dysfunction, then other causes for the patient’s symptoms should be considered
-
If the PASP is >35-40 and/or there is evidence of RV dysfunction AND a common cause is present (left heart disease, high output state, chronic lung disease, sleep apnea, chronic thromboembolic disease), then the suspected common cause should be treated
-
If the PASP is >35-40 and/or there is evidence of RV dysfunction AND a common cause is not clearly present OR a common cause was treated and symptoms persist/worsen, then the patient should be further evaluated with:
-
Invasive hemodynamic testing to confirm PH (mean PAP >= 25), categorize as pulmonary arterial hypertension (PCWP< =15 and PVR > 240 dynes/s/cm5; includes groups 1, 3, 4, 5) or pulmonary venous hypertension (PCWP > 15; group 2), and evaluate prognostic variables (right atrial pressure, cardiac output, pulmonary vascular resistance)
-
Functional testing (most commonly six-minute walk test)
-
Etiologic evaluation as appropriate: lab tests (e.g. HIV, LFTs, ANA), PFTs, VQ scan, sleep testing
-
ECHO findings that may be seen are RA and RV enlargement, reduced RV function, displacement of the interventricular septum with interseptal flattening and D-shaped LV, tricuspid regurgitation (TR), and pericardial effusion.
Echo PASP correlates well with cath PASP, but large discrepancies in the actual numbers (10-20mmHg or more difference between cath and echo) are common in the clinical setting. These discrepancies may be due to invalid assumptions of the equation used to estimate PASP from the tricuspid regurgitation (TR) jet velocity or poor measurement of the TR jet velocity itself.
Mean PAP = 0.61 x PA systolic pressure + 2 mmHg (PA systolic pressure=RVSP)
The diagnosis of PAH requires confirmation with a complete right heart catheterization. The current hemodynamic definition of PAH is a mean pulmonary artery pressure (mPAP) greater than 25 mm Hg; a pulmonary capillary wedge pressure (PCWP), left atrial pressure, or left ventricular end-diastolic pressure (LVEDP) less than or equal to 15 mm Hg.
Treatment:
The initial focus of care should center on addressing any potential reversible causes of acute RV decompensation and development of a strategy to improve RV function. The latter may be achieved through modifying RV preload, contractility, and afterload. Consideration must also be given to maintaining coronary perfusion and avoiding tachycardia. Meticulous fluid management, reducing venous filling pressures, normalizing cardiac output , close monitoring of blood pressure, urine production, RA pressure, and ScvO2 or SvO2 are crucial to guide treatment strategies in these patients.
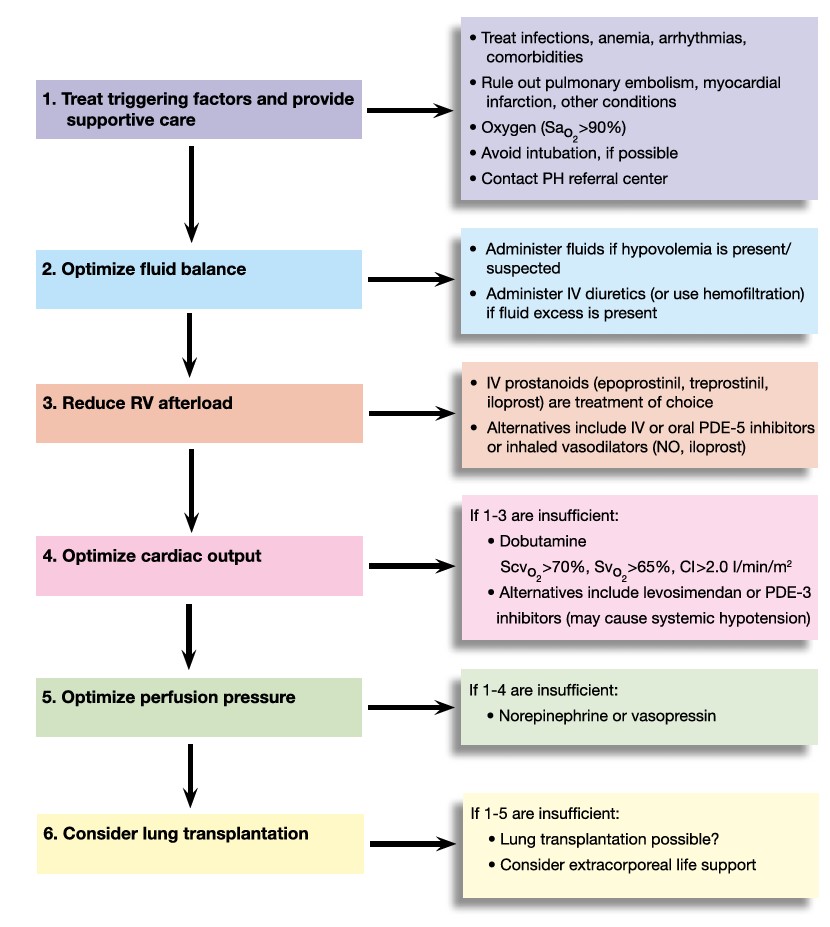
Fluid Management
Fluid management of these patients is often difficult, as both hypovolemia and hypervolemia can have detrimental effects on blood pressure, organ perfusion, and cardiac function. Earlier studies have suggested that fluid loading may improve hemodynamics in patients with acute pulmonary embolism , but unmonitored fluid challenge may further impair RV function. In most, but not all, cases, RV failure is associated with fluid overload and a negative fluid balance is the key to successful therapy. However, fluid removal may reduce the already low cardiac output and may thereby further impair end organ function.
Maintaining Cardiac Output and Systemic Blood Pressure:
Systolic RV failure with low cardiac output and hypotension is more difficult to treat and may require catecholamines or vasopressin to stabilize blood pressure and cardiac output. The b 1 -agonist dobutamine augments myocardial contractility and reduces right and left ventricular afterload, which makes it the preferred catecholamine for patients with RV failure. However, the use of b-adrenergic agents may lead to tachycardia. Patients with pulmonary hypertension may be particularly vulnerable to the adverse effects of tachycardia on diastolic filling time. Consequently, agents that do not have chronotropic properties, such as PDE-3 inhibitors ( Milrinone) may be preferable in some patients. PDE-3 inhibitors may have direct inotropic effects by increasing levels of endogenous cAMP and indirectly augment cardiac function by reducing afterload.
Profound or persistent hypotension, especially in patients with low systemic vascular resistance due to infection, may require additional therapy with norepinephrine, a vasoconstrictor stimulating a 1 – and b 1 -adrenergic receptors. Adequate systemic blood pressure is required to ensure adequate coronary perfusion in these patients, which is a prerequisite to maintain cardiac function. The downside of higher doses of norepinephrine is its potential detrimental effect on pulmonary vascular resistance . Vasopressin may be an alternative to norepinephrine as this drug has systemic vasoconstrictive but pulmonary vasodilatory properties. Levosimendan, a calcium sensitizing agent with positive inotropic and vasodilatory effects, holds promise for patients with PH and RV failure.
Oxygen and Ventilator Support
Maintaining sufficient oxygen supply is self-evident. This includes supplemental oxygen to keep oxygen saturation above 90% and the correction of anemia, if present. The ideal hemoglobin level of patients with RV failure due to PH has never been studied, but given the likelihood that anemia may worsen RV function, we suggest that hemoglobin levels greater than 10 g/dl be maintained.
Every attempt should be made to avoid endotracheal intubation of patients with RV failure . Intubation of these patients is often problematic owing to effects of sedatives on cardiac function and nonselective vasodilation leading to systemic hypotension and hemodynamic collapse. Also, positive pressure will increase RV afterload and decrease LV afterload.
Opioids or sedatives should be administered with great care to avoid drops in blood pressure. If intubation and mechanical ventilation are unavoidable, hypotension and loss of RV contractility must be prevented and the administration of catecholamines before anesthesia should be considered. Maintenance of anesthesia is usually achieved with low-dose opioids or ketamine together with benzodiazepines or propofol. Airway pressures should be kept to a minimum while at the same time hypercapnia must be prevented because of its deleterious effects on pulmonary hemodynamics.
Surgical interventions:
Balloon atrioseptostomy (BAS) is used in some pulmonary hypertension centers as treatment of severe pulmonary hypertension. BAS decompresses the enlarged RV and improves LV filling as well as cardiac output. Despite oxygen desaturation, the net effect is usually an increase in systemic oxygen transport.
Reducing RV Afterload
One of the most important interventions to reverse RV failure is to reduce RV afterload through the use of pulmonary vasodilators or PAH-targeted therapies. The importance of reducing RV afterload is emphasized by the rapidity with which RV function is restored after pulmonary endarterectomy and lung transplantation.
There are 3 classes of pulmonary vasodilator drugs: phosphodiesterase-5 inhibitors (PDE-5 inhibitors, e.g. sildenafil, tadalafil), Endothelin receptor antagonists (ERAs, e.g. bosentan, ambrisentan), and prostacyclins (epoprostenol, iloprost, treprostinil). The use of PAH-targeted therapies depends primarily on previous treatment. In therapy-naive patients with PAH (or other severe forms of PH) and RV failure, intravenous prostacyclin derivatives (epoprostenol, treprostinil, iloprost) are the initial treatment of choice, although care must be taken to avoid systemic hypotension. Once these patients have been stabilized, oral therapies with endothelin receptor antagonists and PDE-5 inhibitors may be added with or without later withdrawal of the prostanoid.
The general approach to group 1 patients is to consider anticoagulation, diuretics, oxygen therapy, and digoxin. If patients have a vasodilator response during the catheterization procedure, this predicts long-term response to oral calcium channel blockers and a trial should be initiated and continued if there is a sustained response. If not, the therapeutic approach is guided by functional status (WHO symptom classification) and objective testing (e.g. 6MWT) as follows:
-
Low risk, functional class I-III patients may be treated with oral PDE-5 inhibitors, oral ERAs, or inhaled prostacyclins.
-
High risk, functional class III-IV patients should be treated with intravenous or subcutaneous prostacyclins.
-
Patients should be re-evaluated for improvement on therapy and considered for combination therapy. Clearly, these patients should be managed by clinicians with expertise in PH.
Inhaled vasodilators such as nitric oxide or iloprost might be used as supplementary therapy, especially in patients who do not tolerate parenteral prostanoids because of systemic hypotension. For patients in whom all conventional treatment options including intravenous prostacyclin derivatives have been exhausted, ICU treatment will have to rely primarily on the general measures outlined previously, focusing on careful fluid management and a judicious choice of catecholamines.
Acute vasodilator testing, which involves the administration of pharmacologic agents to test the presence of pulmonary vasoreactivity, has prognositic value and should be performed in all IPAH patients who might be considered potential candidates for long-term calcium-channel blocker therapy. Those with overt right heart failure or hemodynamic instability should not undergo acute vasodilator testing. The definition of an acute responder is a reduction in mPAP of at least 10 mm Hg to an absolute mPAP of less than 40 mm Hg without a decrease in cardiac output.
PH Associated with Left Heart Disease
Patients with systolic left ventricular dysfunction may have an increase in RV afterload due to a passive increase in pulmonary pressure resulting from an increase in left ventricular end diastolic pressure and, variably, an active component relating to an increase in transpulmonary gradient (mean pulmonary arterial pressure – PCWP). The normal transpulmonary gradient is <12 mmHg. In these patients, treatment should focus on strategies to improve left ventricular function through manipulating preload, contractility, and afterload. However, even after successful treatment and a reduction in left atrial/ left ventricular end-diastolic pressure, the transpulmonary gradient may remain elevated. (Note: In PH due to left heart failure, the transpulmonary gradient remains normal. i.e. if PCWP increases, MPAP increases by the same extent).This is particularly relevant for patients who are being considered for heart transplantation or left ventricular assist device placement, where the presence of a refractory elevation in PVR and RV dysfunction may disqualify them for transplantation or lead to a requirement for mechanical RV support.
The use of pulmonary vasodilators could actually worsen pulmonary venous hypertension and pulmonary edema by increasing pulmonary blood flow in the face of elevated left-sided filling pressures. That said, some of these patients will have developed intrinsic pulmonary vascular disease and could theoretically benefit from pulmonary vasodilator therapy. In fact, a small trial suggested potential benefit of PDE-5 inhibitors in patients with heart failure and a preserved ejection fraction if there was a >5 mmHg difference between PA diastolic pressure and PCWP after optimizing volume and blood pressure. ERAs and prostacyclins should not be used.
PH Associated with Lung Disease
Although pulmonary hypertension may complicate ARDS, it is typically not hemodynamically relevant to patients without preexisting PAH and usually does not require specific treatment. In patients with PH in the setting of obstructive, fibrotic, or hypoventilation syndromes, treatment generally centers around correcting hypoxemia and hypercapnia. Patients presenting with hypercapnic respiratory failure and PH with signs of RV failure often recover rapidly on standard therapy including noninvasive ventilation. Pulmonary vasodilators do not have a role and in fact may worsen VQ matching in these patients, precipitating worsened oxygenation and symptoms.
Chronic Thromboembolic Pulmonary Hypertension
Patients with CTEPH presenting with RV failure are generally treated according to the same principles outlined previously. One important consideration is the likelihood of a new episode of acute pulmonary embolism. Even small pulmonary emboli may cause hemodynamic deterioration in patients with preexisting pulmonary hypertension. Group 4 PH is potentially curable with thromboendartectomy. There may be a role for pulmonary vasodilators in those who are not surgical candidates.
If patients with CTEPH cannot be stabilized by medical therapy, emergency pulmonary endarterectomy should be considered. Of note, this procedure is different from pulmonary embolectomy and requires surgical expertise that is available only in specialized centers.
Pulmonary Venoocclusive Disease
Pulmonary venoocclusive disease represents a significant challenge. This diagnosis should be suspected in a patient who satisfies the diagnostic criteria for idiopathic pulmonary arterial hypertension but who has computed tomographic evidence of patchy ground-glass opacities, septal lines, pleural effusions, and/or mediastinal adenopathy. The pulmonary capillary wedge pressure is often normal in these patients, as disease relates to an increase in pulmonary venous vascular resistance and not alterations in left atrial compliance. These patients may also have significant hypoxemia from the resultant interstitial and alveolar edema. It is important to recognize that classical pulmonary vasodilators may improve cardiac output but worsen lung edema. Apart from attempts at aggressive diuresis there is no temporizing therapy available for these patients.
Pearls:
-
Patients with pulmonary hypertension, in particular those with pulmonary arterial hypertension (PAH), are at high risk when undergoing anaesthesia and major surgery. The perioperative management can be complicated by hemodynamic instability resulting in severe hypoxemia, acute right heart failure/circulatory collapse and death.
-
Patients with PAH are at increased risk for intrapulmonary thrombosis and thromboembolism, due to sluggish pulmonary blood flow, dilated right heart chambers, venous stasis, and a sedentary lifestyle. Warfarin anticoagulation is recommended in all patients with IPAH based on 1 prospective and 2 retrospective observational, uncontrolled trials.
-
Digoxin may be considered in patients with PH who develop atrial tachyarrhythmias.
-
Calcium channel blockers are indicated only for patients who have a positive acute vasodilator response. Long acting nifedipine, diltiazem, or amlodipine are the most commonly used calcium channel blockers. Due to its potential negative inotropic effects, verapamil should be avoided.
-
Continuous intravenous epoprostenol improves exercise capacity, hemodynamics, and survival in IPAH and is the preferred treatment option for the most critically ill patients. Common side effects include headache, jaw pain, flushing, nausea, diarrhea, skin rash, and musculoskeletal pain.
-
Treprostinil, a prostanoid, may be delivered via either continuous intravenous or subcutaneous infusion.
-
Iloprost is a prostanoid delivered by an adaptive aerosolized device 6 times daily.
-
When on prostanoids, Liver function tests must be monitored indefinitely on a monthly basis.
-
(PDE)-5 inhibitors such as Sildenafil and Tadalafil also improve Phosphodiesterase exercise capacity and hemodynamics in PAH. Side effects include headache, flushing, dyspepsia, and epistaxis.
-
Inhaled NO can be a reasonable option and sometimes a first choice as well.
-
Endothelin receptor antagonists such as Busentan and Ambrisentan are gaining popularity. In addition to potential hepatotoxicity, other side effects include anemia and the development of edema.
-
Lung transplant is an option in patients who failed everything else. ECMO can be considered as a bridge, if patient is on transplant list.
-
Lifelong anticoagulation is indicated in chronic thrombo-embolic pulmonary hypertension.
-
Patients with non-PAH pulmonary hypertension can be treated with PAH specific therapy, provided the underlying disease is adequately treated. For example, treat left heart failure/ COPD/ sleep apnea first before considering PAH specific therapy.
-
Half life of inhaled NO is 15-30 secs and duration of action is 5 mins
-
Half life if i.v Flolan is 3-5 mins. Abrupt interruption of the epoprostenol infusion should be avoided as, in some patients, this may lead to a rebound PH with symptomatic deterioration and even death.
-
Pregnancy is associated with 50% mortality in patients with PH.
-
Elective surgeries should be deferred until PH is controlled as general anesthesia can have deleterious effects on hemodynamics.
-
Liver tests needs to be monitored on bosentan therapy.
-